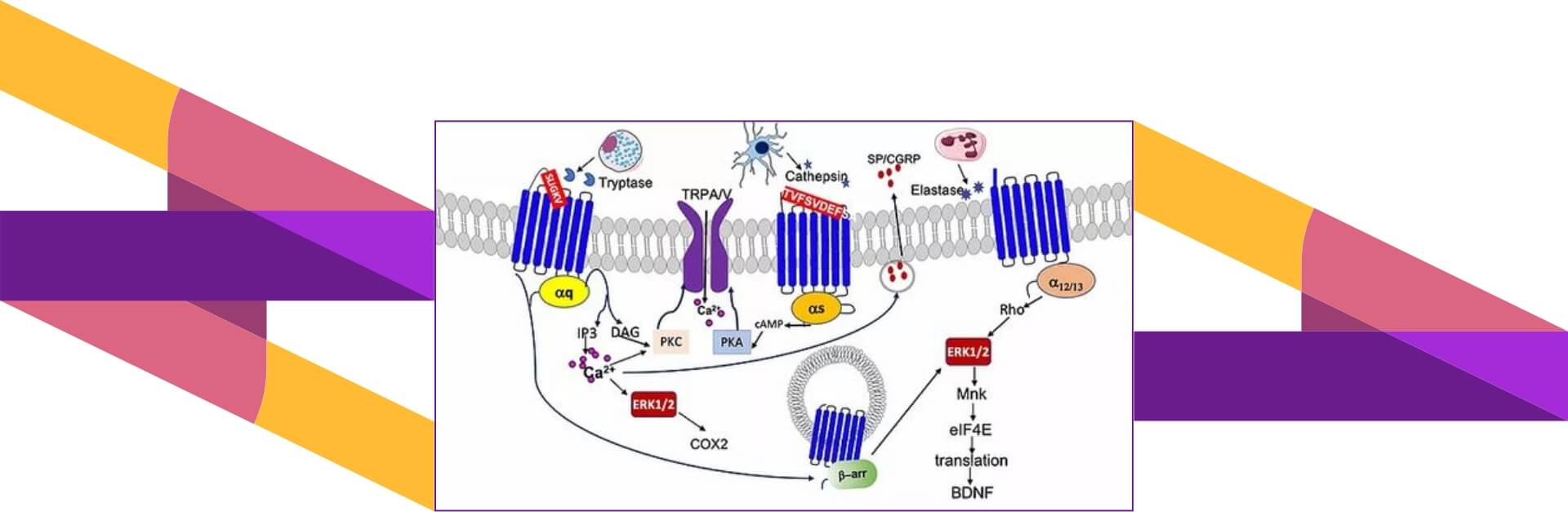
How does PAR2 work therapeutically?
PAR2 is widely expressed throughout the body-in neurons and immune cells, in the epithelial cells of the respiratory and gastrointestinal system, in the vasculature and in the skin. It can be activated by many different proteases, that cleave it within at specific sites within the extracellular N-terminus. PAR2 was first identified as a receptor for trypsin, which is released from epithelial cells in the GI tract and airway.
We now know it can be activated by a number of trypsin-like proteases, such as tryptase, released from mast cells, membrane associated proteases such as TMPRSS2 that is expressed in airway epithelial cells, and proteases released from allergic pathogens such as AASP (Alternaria Alternata Serine Proteinase). Other proteases (Cathepsin-S, Elastase, and Kallikreins) can also activate PAR2. PAR2 signaling is dependent upon the activating protease.